Attosecond electron dynamics in molecules
One of the visions guiding current research on the application of attosecond (1 as = 10-18s) laser pulses to molecules is the possibility of controlling molecular processes by manipulating valence electrons on attosecond to few femtosecond (1 fs = 10-15s) timescales. As predicted by several theoretical works in the last decade, the ultrafast removal (on attosecond timescales) of electrons from molecules may lead to ultrafast migration of the hole in the charge density along the molecular skeleton, potentially influencing the subsequent chemistry of the molecule in what is sometimes called “charge-directed reactivity”. An important and necessary challenge that needs to be overcome in order to observe charge migration is to prove that attosecond pulses can observe this process. In a study that was published in Physical Review Letters, experimental results are published that were obtained in the new attosecond laboratories at the Max-Born-Institute and that provide the first evidence for this.
Attosecond laser pulses are the shortest laser pulses that scientists are able to produce. Using a process called high-order harmonic generation (HHG), it has become possible to produce pulses with a duration of 50-500 as. In HHG, an atomic or molecular gas is typically exposed to a near-infrared driving laser pulse that is intense enough to ionize the gas. The electrons that are thereby set free experience the oscillatory electric field of the driving laser, which pulls them away from their parent ion, and then, a short time later (when the laser electric field has changed sign), accelerates them back towards this ion. In the course of the electron-ion re-collision that then occurs, the electron may be re-absorbed and all the energy that has been invested in the ionization and laser acceleration of the electron may come available via the formation of high energy photons, with an energy that is typically in the 10-100 eV range. Since all electrons that participate in the process are set free and recombine more or less simultaneously, these bursts of high energy photons have a duration that is merely a small fraction of the duration of one optical cycle of the driving laser. Since this duration is just a few femtoseconds for a near-infrared driving laser pulse, the formation of attosecond pulses is inevitable.
Attosecond pulses are a wonderful tool for studying the time-dependent motion of electrons. Whereas the motion of atomic centers is most appropriately studied with femtosecond laser pulses, making movies of electronic motion requires a time resolution in the attosecond domain. Therefore, since their discovery in 2001, many scientists have turned their attention to the application of attosecond pulses to studies of electron dynamics in atomic, molecular and condensed phase systems. In previous studies by the MBI-team on the application of attosecond pump-probe spectroscopy to a molecular system, first evidence was obtained for coupling of electronic and structural dynamics on attosecond to few-femtosecond timescales, as well as for entanglement of multiple electrons. However, the use of attosecond pulses for observing purely electronic motion in a neutral molecule thus far remained elusive.
The recently published successful observation of electron dynamics in neutral molecules, which were performed in the framework of an international collaboration with researchers from the universities of Lyon (France) and Lund (Sweden), bases itself on the technique of “dynamic alignment” that the MBI-team pioneered a little over a decade ago, and that has become a preparation step for many experiments addressing the structure and time-dependent dynamics in molecules. In a dynamic alignment experiment, a molecule is exposed to a laser pulse that is too weak to ionize the molecule, but strong enough to induce a dipole in the molecule. The interaction of the induced dipole with the laser electric field leads to a situation where the molecules align their most polarizable axis along the laser polarization axis. This allows researchers to determine how processes that are observed in the laboratory relate to processes that occur in the molecular frame.
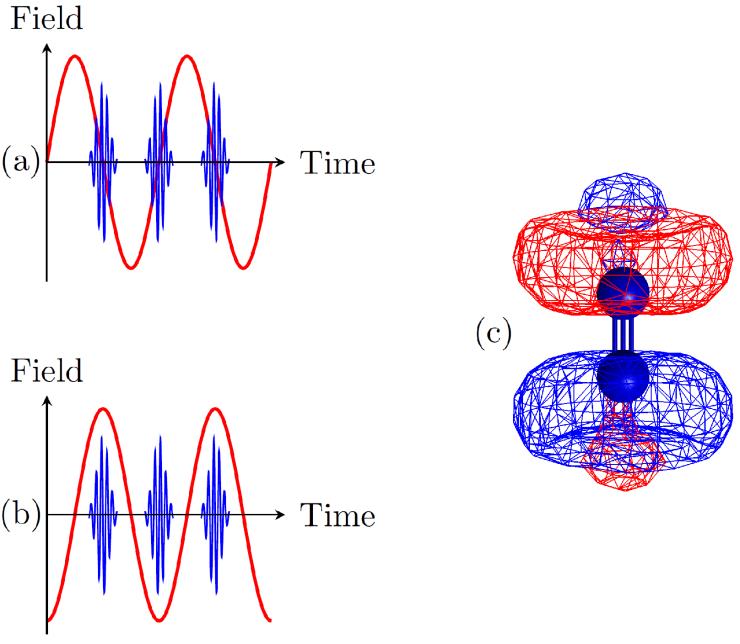
In these recent experiments, the oscillating dipole that causes the “dynamic alignment” effect was directly observed, by ionizing the molecule with a train of attosecond laser pulses that was synchronized to the field oscillations of a near-infrared laser that induced the dipole. For a series of molecules, it was observed that the ionization probability by the attosecond pulses was markedly different at times when the oscillatory electric field of the near-infrared laser induced a dipole (Figure 1 (b)) and at times where this dipole was momentarily zero (Figure 1 (a)). The differences in the electron density in these two cases are shown for the case of molecular nitrogen in Figure 1 (c). The reason for the different ionization probabilities is that the absorption of light requires that both energy and momentum need to be conserved, in the case of the high energy photons in the attosecond pulse strongly favoring times when the electron that is to be removed is relatively close to one of the atomic centers.
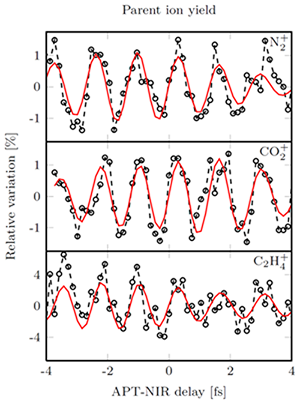
The experiment was performed for a series of molecules (Figure 2), and showed that the observed effect scaled nearly linearly with the polarizability of the molecule under investigation. As described in the publication, the experiment may be viewed as an attosecond timescale implementation of the technique of molecular Stark spectroscopy, where the dependence of molecular photoabsorption on the presence of an external electric field is investigated. In the future, the team will try to measure the absorption spectrum with attosecond time resolution and will try to use the attosecond pulses to observe charge redistribution that is not driven by an external laser, but that is intrinsic to the molecule.
Figure 1: Synchronization of the near-infrared (NIR) field (red line) with the attosecond pulse train (APT, blue line). (a) The APT is synchronized to the zero crossings of the NIR field, while in (b) it is synchronized to the NIR field extrema. In this situation the electron density is different (compared to the undisturbed molecule) when the APT ionizes the molecule. The change of the electron density in the case of molecular nitrogen is plotted in (c). The red color denotes sites of increased density while blue shows the opposite.
Figure 2: The observed variation of the parent ion yield as a function of pulse delay. The molecules (molecular nitrogen, carbon dioxide and ethylene) were under the influence of a NIR field when ionized by an APT. The modulation increases with increasing polarizibility. The result of Fourier filtering around the double frequency of the NIR field is indicated by the red line.
Link: http://prl.aps.org/abstract/PRL/v111/i3/e033001
Contact:
Max Born Institute for Nonlinear Optics and Short Pulse Spectroscopy (MBI)
Prof. Marc Vrakking
Tel.: +49-30-6392-1200
Christian Neidel
Tel.: +49-30-6392-1238